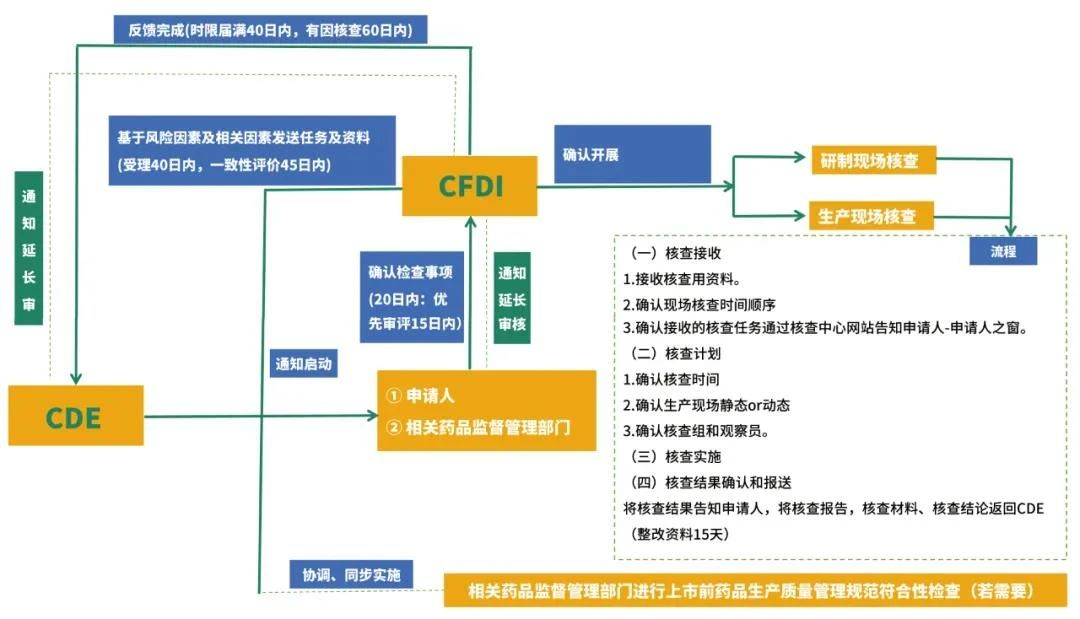
药品注册申请受理后,药品审评中心(CDE)在药品注册申请受理四十日内通知药品核查中心(CFDI)启动注册核查工作,以核实申报资料的真实性、一致性以及药品上市商业化生产条件,检查药品研制的合规性、数据可靠性等。
注册现场核查流程图
备注:
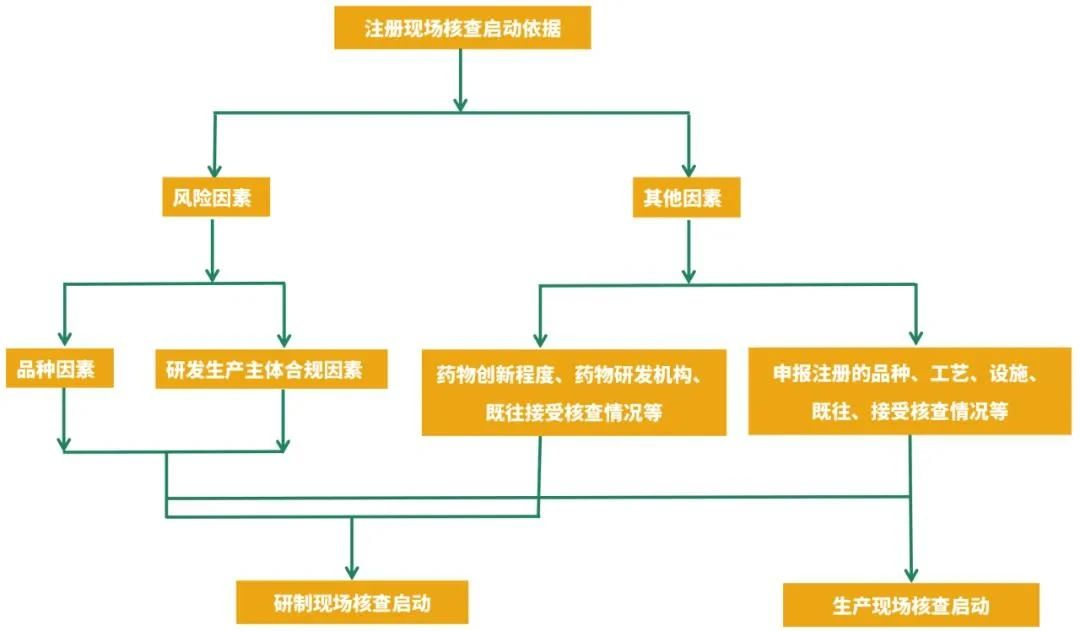
核查启动因素示意图


备注:
原则上以品种因素和研发生产主体合规因素中风险情形较高的确定注册申请风险等级,特殊情况的除外。对于补充申请,生物制品和中药注射剂不纳入品种因素高风险情形。
研制现场核查内容
药品研制基本情况(如属委托,应说明被委托研究单位基本情况)。
研制所涉及的批次(含BE批)批量用途,研制批次(含BE批)生产的地址、生产线、时间地点、使用量和剩余量等。
研制批次(含BE批)所用处方生产工艺、原辅料包装材料来源及标准、生产线(设备设施)、产品质量标准(含中间控制标准)等是否与已上市/拟上市商业化生产规模的批次一致。
参比制剂的来源、采购和使用情况。
药品和参比制剂体外研究的对比研究情况,研究时间、批号和研究结果。(质量对比和溶出曲线)
药品关键质量属性(含稳定性)研究情况。(全检数据及稳定性数据)
接受现场检查品种全套注册申报资料。
委托研究协议和质量协议,如有。
参比制剂的来源及证明,如购买发票、赠送证明等。参比制剂的包装标签、说明书、剩余样品等。参比制剂的接收、发放、使用记录或凭证。
药品相关研究记录,包括:处方工艺研究原始记录,如有;样品试制相关原始记录;质量研究相关原始记录;体外评价及稳定性研究的相关原始记录;仪器设备使用记录;纸质图谱及电子图谱。
其他相关文件,包括:处药品检验方法确认或验证资料;稳定性试验方案及报告;体外研究总结报告;溶出度仪的验证资料;研究用的剩余样品情况(不应销毁)。